Oprócz doskonałego opisu konstruktywnej i destrukcyjnej ingerencji w powstawanie mikroorganizmów @ Nanoputian, chcę przedstawić bardziej matematyczne wyjaśnienie, dlaczego faza funkcji falowej nie ma znaczenia.
Znajdowanie funkcji falowej
Niezależne od czasu równanie Schrödingera, w jednym wymiarze, brzmi:
$$ \ hat {H} \ psi (x) = E \ psi (x) $ $
Można wykazać, że jeśli funkcja falowa $ \ psi = \ psi (x) $ spełnia powyższe równanie, funkcja falowa $ k \ psi $ (z $ k \ in \ mathbb {C} $) spełnia również powyższe równanie z tą samą wartością własną energii $ E $. Jest to spowodowane liniowością hamiltonianu:
$$ \ begin {align} \ hat {H} (k \ psi) & = k (\ hat {H} \ psi) \\ & = k (E \ psi) \\ & = E (k \ psi) \ end {align} $$
Jest kilka warunków, które musi spełniać funkcja falowa aby była fizycznie możliwa do zrealizowania, tj. reprezentowała „prawdziwą” fizyczną cząstkę. W tej dyskusji istotnym warunkiem jest to, że funkcja falowa musi być integrowalna z kwadratem (lub normalna ). W kategoriach matematycznych:
$$ \ langle \ psi \ lvert \ psi \ rangle = \ int _ {- \ infty} ^ {\ infty} \! \ Lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x < \ infty $$
Oznacza to, że musi istnieć stała $ N \ in \ mathbb {C} $ taka, że $ N \ psi $ jest znormalizowana :
$$ \ int _ {- \ infty} ^ {\ infty} \! \ lvert N \ psi \ rvert ^ 2 \, \ mathrm {d} x = \ lvert N \ rvert ^ 2 \! \! \ int _ {- \ infty} ^ {\ infty} \! \ lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x = 1 $$
Od tego momentu, założymy, że już znaleźliśmy odpowiednią stałą normalizacyjną taką, że funkcja falowa $ \ psi $ jest już znormalizowana. Innymi słowy, załóżmy, że $ \ langle \ psi \ lvert \ psi \ rangle = 1 $, ponieważ możemy. Rozważmy teraz funkcję falową $ - \ psi $, która jest równoważna $ N \ psi $ z $ N = -1 $. Czy ta nowa funkcja falowa jest znormalizowana?
$$ \ begin {align}
\ int _ {- \ infty} ^ {\ infty} \! \ lvert - \ psi \ rvert ^ 2 \, \ mathrm {d} x & = \ lvert -1 \ rvert ^ 2 \! \! \ int _ {- \ infty} ^ {\ infty} \! \ lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x \\ & = \ int _ {- \ infty} ^ {\ infty} \! \ lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x \\ & = 1 \ end {align} $$
Oczywiście. Tak więc to, co napisałem do tej pory, zasadniczo mówi: jeśli $ \ psi $ jest znormalizowanym rozwiązaniem równania Schrödingera, więc jest $ - \ psi $ .
W rzeczywistości , możesz pójść o krok dalej. Używając dokładnie tego samego działania, co powyżej, możesz pokazać, że jeśli $ \ psi $ jest znormalizowanym rozwiązaniem równania Schrödingera, funkcja falowa $ (a + ib) \ psi $ również będzie równa jeden, o ile $ a ^ 2 + b ^ 2 = 1 $. (Jeśli lubisz wykładniki, jest to równoznaczne z powiedzeniem $ a + ib = e ^ {i \ theta} $.) Zilustrowałem ten pomysł na poniższym diagramie:
$ \ qquad \ qquad \ qquad \ qquad \ qquad \ qquad $ 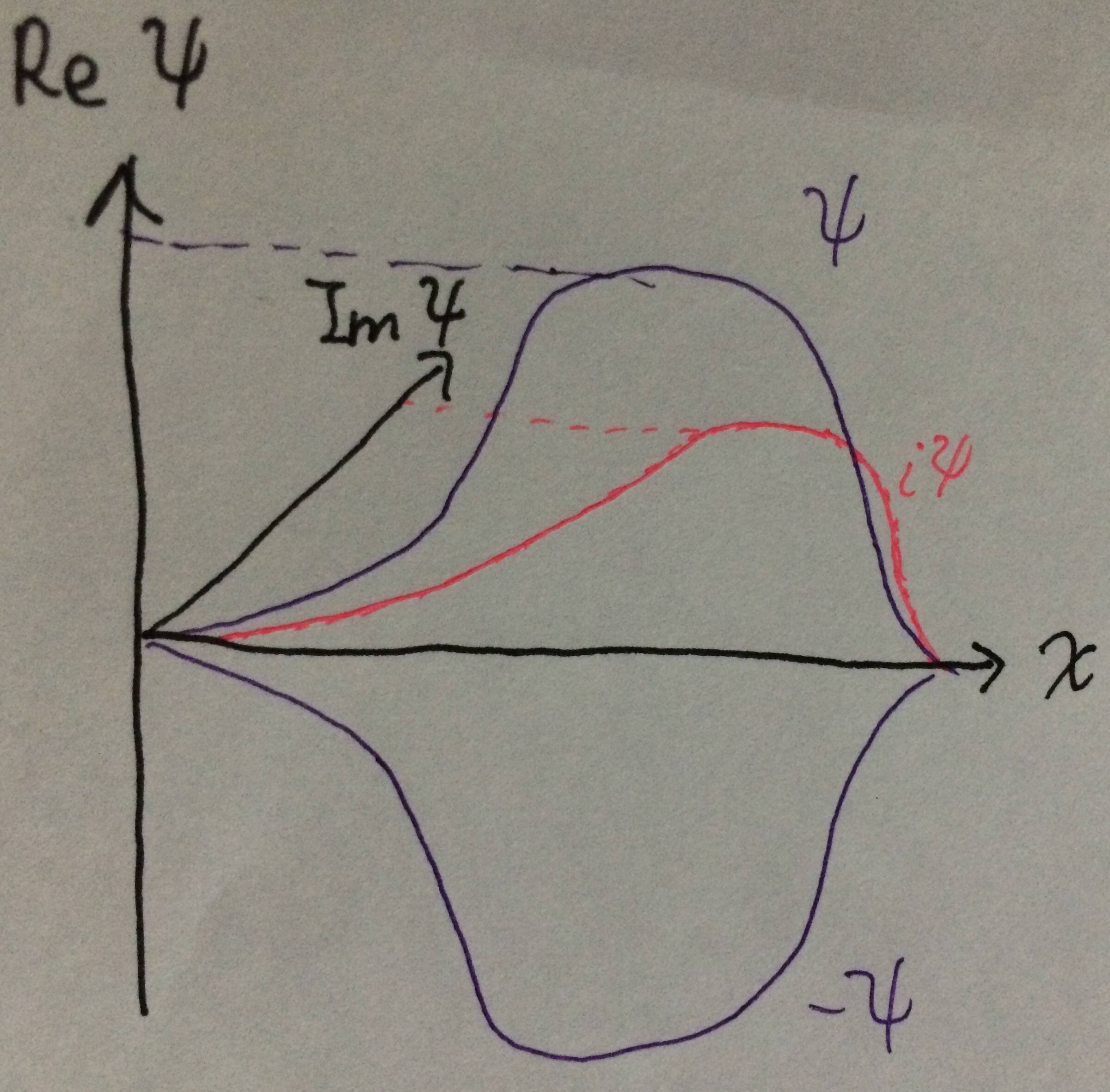
Jeśli $ \ psi $ jest jednowymiarową funkcją falową o wartości rzeczywistej, możesz wykreślić ją na wykresie względem $ x $. Funkcja falowa $ i \ psi $ miałaby wtedy dokładnie ten sam kształt, po prostu wychodząc z płaszczyzny papieru ($ \ theta = 90 ^ \ circ $). Mogłabyś mieć funkcję falową $ (1 + i) \ psi / \ sqrt {2} $. Byłby skierowany na zewnątrz płaszczyzny papieru o $ \ theta = 45 ^ \ circ $, dokładnie w połowie odległości między $ \ psi $ a $ i \ psi $, ale dokładnie w tym samym kształcie. Jednak fizyka nie wie, gdzie jest płaszczyzna twojego papieru, więc wszystkie te funkcje falowe są jednakowo dopuszczalne. Z punktu widzenia systemu wszystkie są tym samym.
Korzystanie z funkcji falowej
„Ale czekaj! Jeśli funkcja falowa jest ujemna, co o wartościach pędu, pozycji i energii, które obliczasz? Czy staną się ujemne? ”
„ Dobre pytanie, ja! ”
Cóż, na początek, jedną rzeczą, do której używasz funkcji falowej, jest znalezienie gęstości prawdopodobieństwa, $ P (x) $. Zgodnie z interpretacją funkcji falowej Maxa Borna, jest ona określona wzorem $ P (x) = \ lvert \ psi \ rvert ^ 2 $. Powiedzmy, że gęstość prawdopodobieństwa opisana przez ujemną funkcję falową $ - \ psi $ jest inną funkcją $ x $, nazywaną $ Q (x) $:
$$ \ begin {align} Q (x ) = \ lvert - \ psi \ rvert ^ 2 & = \ lvert -1 \ rvert ^ 2 \ lvert \ psi \ rvert ^ 2 \\ & = \ lvert \ psi \ rvert ^ 2 \\ & = P (x) \ end {align} $$
Zatem gęstość prawdopodobieństwa opisana przez ujemną funkcję falową jest dokładnie taka sama. W rzeczywistości gęstość prawdopodobieństwa opisana przez $ i \ psi $ jest dokładnie taka sama.
Porozmawiajmy teraz o obserwowalnych , takich jak pozycja $ x $, pęd $ p $ i energia $ E $. Każde obserwowalne ma swój operator: odpowiednio $ \ hat {x} $, $ \ hat {p} $ i $ \ hat {H} $ (Hamiltonian ma specjalną literę, ponieważ jego nazwa pochodzi od Williama Hamiltona). Używasz tych operatorów do obliczania średniej wartości obserwowalnych. Podam przykład dotyczący pędu. Jeśli chcesz znaleźć średni moment pędu oznaczony $ \ langle p \ rangle $, wykonaj następujące czynności:
$$ \ begin {align} \ langle p \ rangle & = \ langle \ psi \ lvert \ hat {p} \ rvert \ psi \ rangle \\ & = \ int _ {- \ infty} ^ \ infty \! \ psi ^ * \ hat {p} \ psi \, \ mathrm {d} x \ end { align} $$
Nazwę wartość tej całki $ p_1 $. Teraz zróbmy to samo. Załóżmy, że średni pęd ujemnej funkcji falowej niekoniecznie ma taką samą wartość. Nazwijmy nowy średni pęd czymś innym, na przykład $ p_2 $.
Zanim przejdziemy dalej, ustalę, że operator pędu $ \ hat {p} = -i \ hbar \ frac {\ mathrm {d}} {\ mathrm {d} x} $ jest również liniowy . Jeśli w to wątpisz, możesz to sprawdzić, używając definicji liniowości w pierwszym linku, który zamieściłem. W rzeczywistości wszystkie operatory mechaniki kwantowej odpowiadające obserwablom są liniowe. Dlatego $ \ hat {p} (- \ psi) = - \ hat {p} \ psi $ i tak:
$$ \ begin {align} p_2 & = \ langle - \ psi \ lvert \ hat {p} \ lvert- \ psi \ rangle \\ & = \ int _ {- \ infty} ^ \ infty \! (- \ psi) ^ * \ hat {p} (- \ psi) \, \ mathrm {d} x \\ & = (-1) ^ 2 \! \! \ int _ {- \ infty} ^ \ infty \ ! \ psi ^ * \ hat {p} \ psi \, \ mathrm {d} x \\ & = \ int _ {- \ infty} ^ \ infty \! \ psi ^ * \ hat {p} \ psi \, \ mathrm {d} x \\ & = p_1 \ end {align} $$
A więc jeśli mówimy o stanie podstawowym cząstki w pudełku o długości $ L $, niezależnie od tego, czy używasz dodatniej funkcji falowej
$$ \ psi_1 = \ sqrt {\ frac {2} {L}} \ sin {\ left (\ frac { \ pi x} {L} \ right)} $$
lub ujemna funkcja falowa
$$ - \ psi_1 = - \ sqrt {\ frac {2} {L}} \ sin {\ left (\ frac {\ pi x} {L} \ right)} $$
lub złożona funkcja falowa
$$ i \ psi_1 = i \ sqrt { \ frac {2} {L}} \ sin {\ left (\ frac {\ pi x} {L} \ right)} $$
otrzymasz dokładnie takie same wartości dla średniej pozycji $ (= L / 2) $, średni pęd $ (= 0) $ i średnia energia $ (= h ^ 2 / 2mL ^ 2) $ (słowo średnia jest tutaj zbędne, ponieważ jest to stan stacjonarny, ale cokolwiek) .
Wszystko, co powiedziałem do tej pory, można łatwo uogólnić na trzy wymiary. Można go również uogólnić na liniowe kombinacje stanów stacjonarnych, czyli rozwiązania zależnego od czasu równania Schrödingera.
Uwaga o orbitali molekularnych
„OK, ale co się stanie, gdy połączysz orbitale atomowe, aby utworzyć orbitale molekularne? Masz konstruktywną interferencję z dodatnim + dodatnim i destrukcyjną interferencją z dodatniego + ujemnego, ale co z kombinacją ujemnego + ujemnego?”
„Dobre pytanie, ja!”
Porozmawiajmy o cząsteczce $ \ ce {H2} $. Właściwym sposobem znalezienia orbitali molekularnych jest rozwiązanie równania Schrödingera dla całego układu, co jest naprawdę trudne. Jednym ze sposobów znalezienia przybliżonych form mikroorganizmów jest tworzenie liniowych kombinacji orbitali atomowych; ta metoda nazywa się przybliżeniem LCAO . Nazwijmy orbital 1s wodoru po lewej stronie $ \ phi_1 $ i orbital 1s wodoru po prawej stronie $ \ phi_2 $. Z poprzednich części ustaliliśmy już, że jeśli chodzi o atom wodoru, poszczególne fazy $ \ phi_1 $ i $ \ phi_2 $ nie mają znaczenia . Więc załóżmy dla uproszczenia, że obie ich fazy są pozytywne.
Teraz, z tego, co już wiesz, możesz otrzymać dwa orbitale molekularne $ \ psi_1 $ i $ \ psi_2 $:
$$ \ begin {align} \ psi_1 & = \ phi_1 + \ phi_2 \\\ psi_2 & = \ phi_1 - \ phi_2 \ end {align} $$
To są orbitale wiążące i antybakteryjne odpowiednio (przynajmniej w ramach stałej normalizacji, na której mi się tutaj nie zależy, bo szczegóły są nieistotne). Porozmawiajmy teraz o tych kombinacjach, których przegapiliśmy.
$$ \ begin {align} - \ phi_1 - \ phi_2 & = - \ psi_1 \\ - \ phi_1 + \ phi_2 & = - \ psi_2 \ end {align} $$
Powiedzieliśmy już, że $ \ psi_1 $ i $ \ psi_2 $ są (przybliżeniami) rozwiązaniami równania Schrödingera. Oznacza to, że z tego, o czym mówiliśmy wcześniej, $ - \ psi_1 $ i $ - \ psi_2 $ również muszą być (przybliżeniami) rozwiązaniami równania Schrödingera. Muszą mieć te same energie co $ \ psi_1 $ i $ \ psi_2 $. W rzeczywistości, o ile cząsteczka wie (i dba o to), są one tym samym , co $ \ psi_1 $ i $ \ psi_2 $.
Teraz, ponieważ poszczególne fazy orbitali atomowych nie mają znaczenia, gdybyś naprawdę chciał, mógłbyś ogłosić całemu światu, który zdefiniujesz:
$$ \ phi_3 = \ phi_1 \ tekst {i} \ phi_4 = - \ phi_2 $$
tj. lewy orbital wodoru 1s, $ \ phi_3 $, jest dodatni, a prawy orbital wodoru 1s, $ \ phi_4 $, jest ujemny. W takim przypadku możesz skonstruować orbitale molekularne:
$$ \ begin {align} \ psi_1 & = \ phi_3 - \ phi_4 \\\ psi_2 & = \ phi_3 + \ phi_4 \ end {align} $$
Współczynniki orbitali atomowych musiałyby być różne, ponieważ nalegałeś na umieszczenie ich w różnych fazach - jednak wynik jest taki sam! Otrzymujesz jeden MO wiążący i jeden MO przeciwdziałający.