Normalnym sposobem rysowania profilu reakcji jest umieszczenie bariery między reagentami a produktami, tak jak pokazano w pytaniu. Jako szczyt bariery jest stan przejściowy . Ścieżka reakcji zwykle nie jest dobrze zdefiniowana jako taka, ponieważ istnieje wiele kątów wiązania i odległości, które zmieniają się podczas reakcji, niemniej jednak ta koncepcja ścieżek jest bardzo przydatna w zrozumieniu tego, co się dzieje. To, co pokazuje rysunek, a także ładna animacja, to to, że reagenty zbliżają się, występuje jakaś interakcja, a następnie powstają produkty. Jednak obraz (lub animacja) nie mówi nam, jak to się dzieje i jakie są ważne czynniki, pozostawia się wyobraźni.
Po pierwsze, reagenty muszą uzyskać wystarczającą ilość energii, aby osiągnąć stan przejściowy na szczycie bariery. Średnia energia reagentów jest zwykle przedstawiana jako linia po lewej stronie rysunku ze słowem reagenty. Ta energia jest zwykle znacznie mniejsza niż wysokość bariery (z wyjątkiem reakcji kontrolowanych dyfuzją, patrz poniżej), a to oznacza, że w większości przypadków zderzają się reagenty nie zachodzi reakcja. Po prostu nie ma wystarczającej ilości energii, aby zareagować, niezależnie od tego, czy reakcja jest endotermiczna czy egzotermiczna.
Teraz, ponieważ cząsteczka znajduje się w roztworze lub w fazie gazowej pod pewnym rozsądnym ciśnieniem obojętnego gazu buforowego, wiele zderzeń na sekundę będzie miało miejsce między reagentami a rozpuszczalnikiem (lub gazem buforowym. co najmniej 10 ^ {12} s ^ {- 1} $. Tylko od czasu do czasu, być może 1 na 10 ^ 9 $ zderzeń, będzie wystarczająco energetyczny, aby dać reagentom energię wystarczającą do dotarcia do szczytu bariery. stan przejściowy reakcja może wystąpić, pod warunkiem, że reagenty są w prawidłowej orientacji i zderzenie następuje wystarczająco szybko, aby usunąć wystarczającą ilość energii ze stanu przejściowego, a tym samym obniżyć energię, uniemożliwiając reakcję.
Chociaż średnia energia kinetyczna rozpuszczalnika wynosi 3 $ RT / 2 $ na mol, a < 4kJ / mol jest mała, istnieje rozkład energii podany przez rozkład Boltzmanna. Szansa posiadania energii $ E $ jest proporcjonalna do $ exp (-E / (RT)) $, więc jest bardzo mała, jeśli $ E $ wynosi, powiedzmy, 100 kJ / mol i dlatego wiele reakcji ma małe stałe szybkości i dlaczego tak wiele zderzeń jest potrzebnych, zanim zajdzie reakcja.
Po osiągnięciu stanu przejściowego, ponieważ energia jest tutaj znacznie powyżej średniej, to średnio zderzenie z rozpuszczalnikiem usuwa trochę energii, a nie dodaje do niej, iw ten sposób produkt stabilizuje się.
(Podany tutaj opis jest raczej prymitywny, bardziej szczegółowe rozważania prowadzą do modyfikacji Kramersa w teorii stanu przejściowego, a to obejmuje rozważenie `` tarcia '' na współrzędnej reakcji, zarówno od rozpuszczalnika, jak i od drgań wewnętrznych w reagenty utrudniają lub wspierają ruch wzdłuż współrzędnej reakcji.)
W niektórych reakcjach bariera dla reakcji jest bardzo mała, co oznacza, że jest tylko nieznacznie większa niż średnia energia w tej temperaturze. W tym przypadku, gdy reagenty zderzają się ze sobą, następuje reakcja. Szybkość reakcji jest obecnie ograniczona przez to, jak szybko reagenty mogą się zderzać, a jest to określone przez ich stałe dyfuzji, które z kolei zależą od lepkości roztworu. Są to reakcje „kontrolowane dyfuzją”. W tym samym rozpuszczalniku zawsze mają większą stałą szybkości niż reakcja z większą barierą.
Ważne jest, aby zdać sobie sprawę, że eksperymentalny dowód na to, jakie są stany przejściowe, jest rzeczywiście bardzo, bardzo szkicowy. Na ładnej animacji widzimy, jak wiązania rozciągają się i pękają itp., Ale jest to możliwe, oparte na obliczeniach, z pewnością na intuicji chemicznej i poszlakach z eksperymentów chemicznych, ale które nie są badaniami spektroskopowymi w rozdzielczości czasowej. Przyczyną braku możliwości wykrycia stanu przejściowego jest to, że trwa on najwyżej około 10 ^ {- 13} $, a prawdopodobnie mniej, a nawet w reakcjach zapoczątkowanych femtosekundowym impulsem laserowym nie można ich wykryć nawet w reakcjach fotoindukowanych, takich jak fotoizomeryzacja trans-cis stylbenu (difenyloetenu) przypuszczalnie dlatego, że ich stężenie jest niewielkie.
W prostych reakcjach, takich jak O + H $ _2 $ = H + OH, które są przeprowadzane w wiązce molekularnej, możliwy był znacznie większy postęp. Reakcje te są mierzone przy bardzo niskim ciśnieniu gazu, a wiązka atomów przecina wiązkę cząsteczek, a produkty są analizowane spektroskopowo i / lub pod kątem ich energii i rozkładu przestrzennego.
Możliwe jest wykreślenie powierzchni energii potencjalnej jako serii konturów, tak jak na mapie przedstawiającej wzgórza i doliny. Chociaż powierzchnia jest empiryczna, jest obliczana przy użyciu znanych potencjałów wibracyjnych dla cząsteczek dwuatomowych i metody wiązania Heitler-London Valence. (Powierzchnia nazywa się LEPS od London, Eyring, Polanyi i Sato.)
Rysunek 1 przedstawia wykres energii potencjalnej w funkcji separacji OH + H (wzięty ze środka OH) jako rzędnej, a separację O + H $ _2 $ jako odciętą. (Zwróć uwagę, że w obliczeniach przyjęto, że kolizja jest liniowa (tylko koniec na) między O + H $ _2 $, a obroty H $ _2 $ nie są uwzględniane w ustawianiu początkowych parametrów kolizji) Stan przejścia jest w punkcie około (0,12, 0,09) ), żółte kółko. Jest to „punkt siodła”, w którym powierzchnia ma wypukłą krzywiznę wzdłuż ścieżki reakcji i wklęsłą krzywiznę prostopadłą do niej. Ciemnoniebieski kolor przedstawia najniższą energię, żółto-zielony pośredni, a czerwony najwyższy.
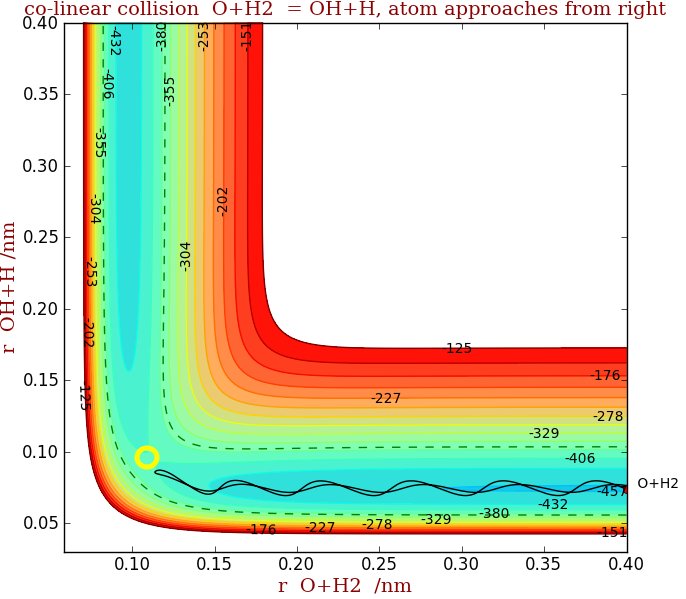
Linia ciągła przedstawia jedną trajektorię liniowa kolizja między O + H $ _2 $ i zaczyna się po prawej stronie figury. Kreskowane linie opisują całkowitą energię, energię zderzenia (kinetyczną) oraz początkową energię drgań w H $ _2 $. Oscylacje w dolinie po prawej stronie pokazują drgania H $ _2 $, gdy zbliżał się on do atomu O. Na rys. 1, chociaż energia całkowita jest wystarczająca, wibracje są takie, że cząsteczka znajduje się w niewłaściwym miejscu na potencjale, ze względu na swoją prędkość początkową i energię wibracji, aby przekroczyć stan przejściowy, a zatem powraca w dół doliny. Nie nastąpiła żadna reakcja.
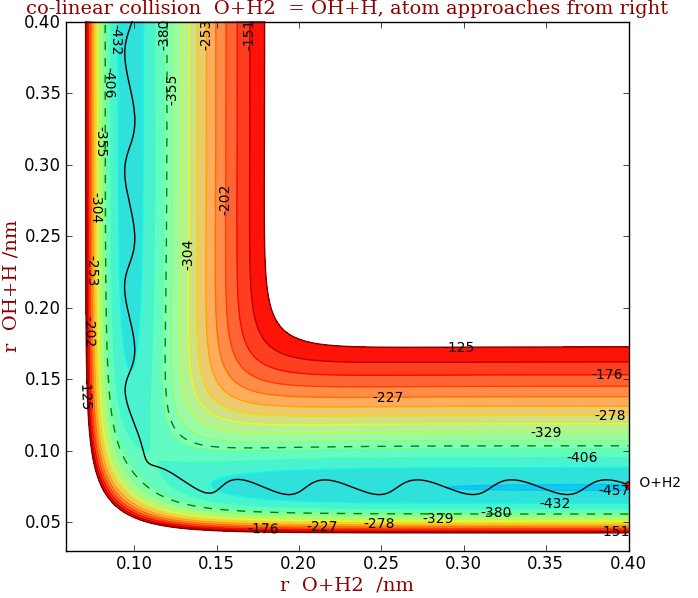
Na drugim rysunku następuje przekroczenie stanu przejściowego, chociaż całkowita energia jest taka sama, ponieważ wibracje umieszczają reagenty we właściwym miejscu na powierzchni. Drgania produktu OH można zobaczyć w pionowej dolinie. Dlatego ważną kwestią jest to, że nie tylko całkowita energia, ale także orientacja i energia wibracji są ważne w wywoływaniu reakcji.
Obserwując energie rotacji i wibracji produktów i reagentów w takich reakcjach, jak te, można ustalić powierzchnię energii potencjalnej. Połączenie ze stałymi szybkości (termicznej) mierzonymi w eksperymentach z normalną kinetyką chemiczną (wiązką niemolekularną) jest dokonywane poprzez obliczenie liczby przejść stanu przejściowego przy danej energii, a następnie uśrednienie energii zderzenia i energii kinetycznej.