Chciałbym poprzeć odpowiedź Klausa niektórymi wynikami z kwantowej teorii atomów w cząsteczkach (QTAIM), opartych na obliczeniach DF-BP86 / def2-SVP. Należy zauważyć, że są to wyniki uzyskane bez uwzględnienia solwatacji lub faz skondensowanych. Sądzę, że nadal dowodzą słuszności w przypadku teorii struktury elektronowej.
Powróciłem do tego pytania, aby odpowiedzieć na inne, podobne pytanie. Wkładając w to więcej wysiłku, zdałem sobie sprawę, że traktowane tutaj struktury są w rzeczywistości stanami przejściowymi. Nie oznacza to, że poruszane kwestie są nieważne. Nawet jeśli istnieją tylko przez bardzo krótkie chwile, nadal istnieją i należy je wziąć pod uwagę.
W o -metyloanilinie można wyraźnie zobaczyć sugerowane wewnątrzcząsteczkowe $ \ ce {H} $ obligacja. Odległość $ \ mathbf {d} (\ ce {NH}) = 239,3 ~ \ mathrm {pm} $ jest tylko trochę mniejsza niż suma promieni van der Waalsa, $ \ mathbf {r} (\ ce {N} ) = 155 ~ \ mathrm {pm} $, $ \ mathbf {r} (\ ce {H}) = 110 ~ \ mathrm {pm} $, ale zaniedbanie tego również jest błędne. Nawet jeśli ta interakcja istnieje tylko przez bardzo krótki czas, nadal oznacza, że stabilizuje ten stan. Nie będzie to jednak cecha dominująca.
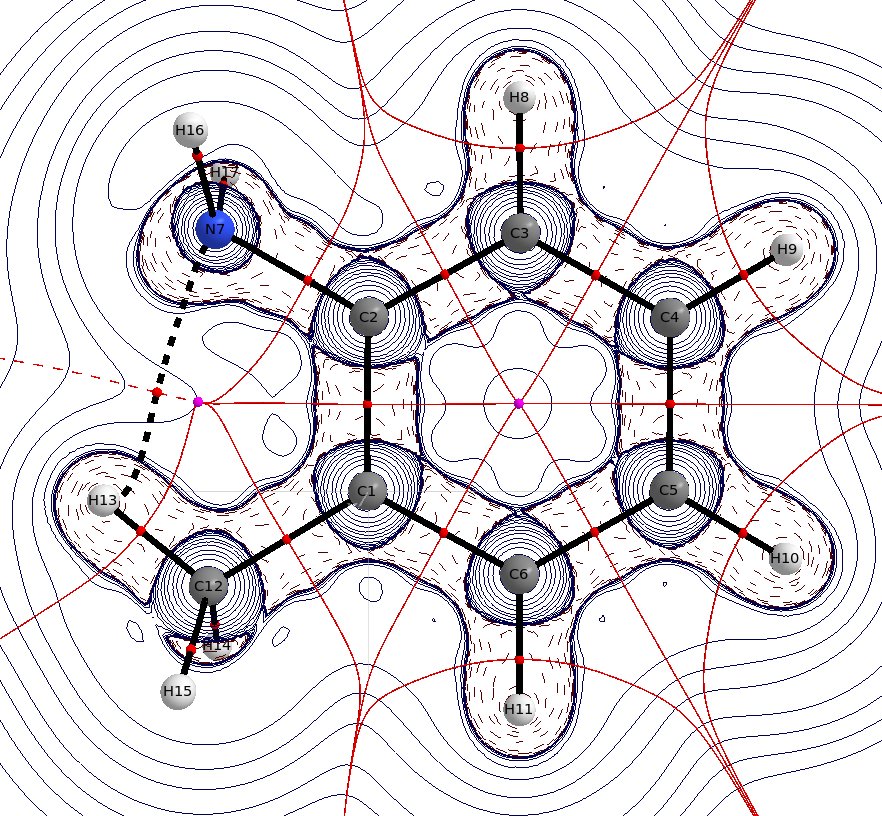
(Rozkład Laplaca, ciągłe niebieskie linie wskazują na wyczerpanie ładunku $ \ nabla ^ 2 \ rho<0 $, przerywane niebieskie linie wskazują akumulację ładunku $ \ nabla ^ 2 \ rho>0 $, Czerwone kule to punkty krytyczne wiązania, fioletowe kule to punkty krytyczne pierścienia, czarne linie to ścieżki wiązania, czerwone linie to powierzchnie o zerowym strumieniu)
Efekty steryczne są zwykle efektami elektronicznymi lub dyspersyjnymi w przebraniu, stąd też odnoszą się do wewnątrzcząsteczkowego wiązania wodorowego. Średnia obligacja $ \ ce {NH} $ wynosi tylko około $ \ mathbf {d} _ \ text {av.} (\ Ce {NH}) \ około 99-105 ~ \ mathrm {pm} $.
Rozwiązanie uwaga użytkownika4604 powinna być nadal brana pod uwagę.
Ta interakcja musi obniżyć powinowactwo do protonów i / lub powinowactwa do kwasu Lewisa, a zatem zmniejsza również zasadowość. Nie tyle dlatego, że jego efekty elektroniczne stabilizują jedną konkretną konformację, ale przede wszystkim sprawiają, że samotna para jest niedostępna przez krótki czas.
Możesz również przeanalizować to pod kątem o -metylobenzoesowy i tutaj efekt zmienia kierunek.
W kwasie benzoesowym jest już pewne wewnątrzcząsteczkowe wiązania wodorowe z orto wodorów, jedno z nich jest nadal obecne w podstawionym przypadku. Odległość $ \ mathbf {d} (\ ce {O-H_ {o '}}) = 226,2 ~ \ mathrm {pm} $ jest tylko trochę mniejsza niż suma promieni van der Waalsa, $ \ mathbf {r} (\ ce {O}) = 151 ~ \ mathrm {pm} $, $ \ mathbf {r} (\ ce {H}) = 110 ~ \ mathrm {pm} $, ale zaniedbanie tego nadal byłoby błędem. Odległość $ \ mathbf {d} (\ ce {O-H_ {Me}}) = 210,8 ~ \ mathrm {pm} $ jest znacznie mniejsza niż suma promieni van der Waalsa. Możesz ponownie zobaczyć interakcję poprzez ścieżkę wiązania i zadzwonić do krytycznych punktów. W zoptymalizowanej geometrii ugrupowanie metylowe jest lekko obrócone, co prowadzi do dwóch równoodległych interakcji. Rotację tej grupy można uznać za swobodną rotację w temperaturze pokojowej.
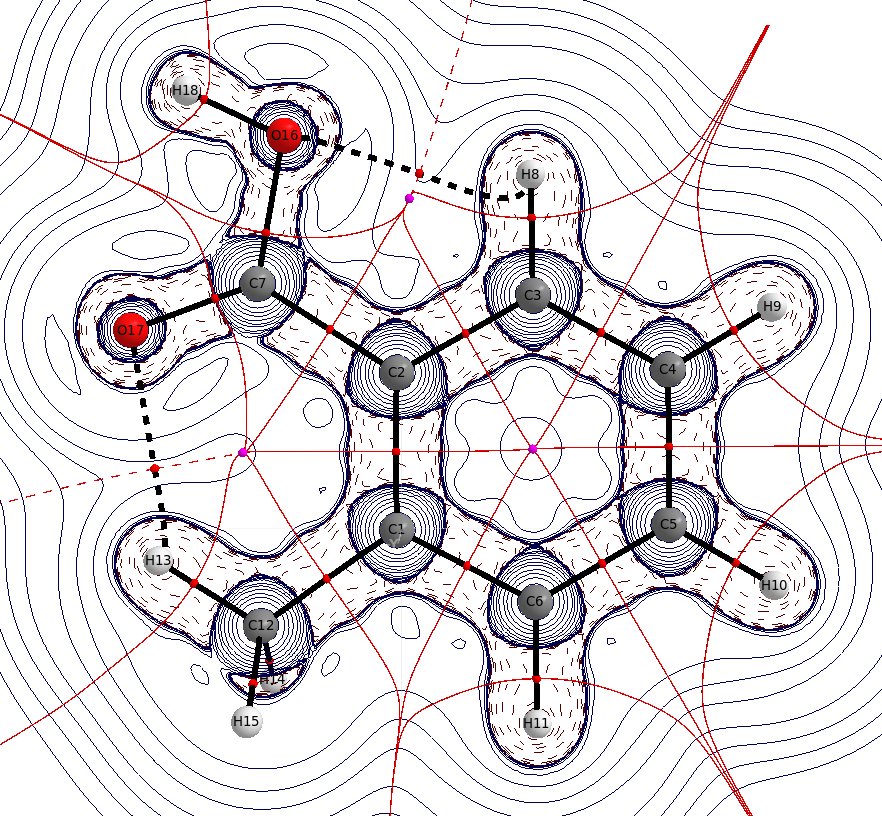
Warto zauważyć, że punkt krytyczny wiązania $ \ ce {O-H_ {kwas}} $ jest prawie w miejscu protonu, co również wskazuje, że większość gęstości elektronów należy już do tlenu.
Jest również oczywiste, że stężenie ładunku w tym wiązaniu jest znacznie niższe niż w jakimkolwiek innym $ \ ce {EX} $ bond, co może wskazywać na słabą więź.